网站导航
13673698348


来自: 发布时间:2022-11-24 浏览 :265次
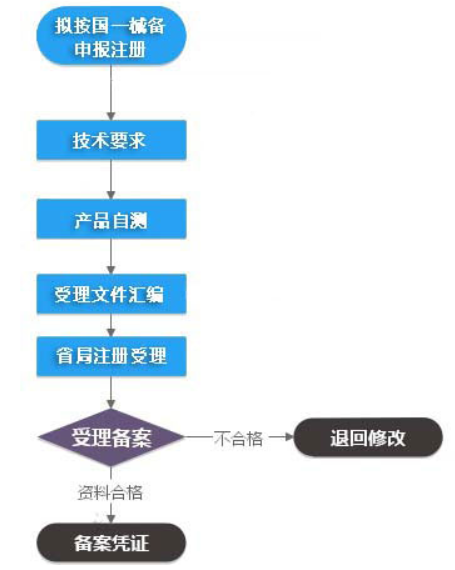
| 要 求 | ||
| 1.产品名称与目录所列内容相同 | ||
| 2.产品描述不超出目录内容 | ||
| 3.预期用途不超出目录内容 | ||
| 4.该事项由本备案单位办理 | ||
| 5.提交了以下九项资料: | ||
| 1)备案表纸质文档已提交。备案表填写完整(不留白) | ||
| 2)安全风险分析报告 | 按要求提交,加盖公章(及骑缝章),写明相关承诺内容。(其中产品检验报告相关填写项不留白) | |
| 3)产品技术要求纸质及电子文档 | ||
| 4)产品检验报告 | ||
| 5)临床评价资料 | ||
| 6)生产制造信息 | ||
| 7)产品说明书及最小销售单元标签设计样稿的纸质及电子文档 | 按要求提交,加盖公章(及骑缝章),提交相关承诺。 | |
| 8)证明性文件 |
提交有效期内的企业营业执照复印件,校验原件、企业名称、地址与备案表一致、加盖公章、加盖“内容与原件一致”的章或手写并签字确认。 | |
| 提交有效期内的组织机构代码证复印件,校验原件、企业名称、机构代码号与备案表一致、加盖公章、加盖“内容与原件一致”的章或手写并签字确认。 | ||
| 提交备案人委托相关人员办理第一类医疗器械备案变更事务的委托书,加盖公章。 提交被委托人身份证复印件,校验原件、加盖公章、加盖“内容与原件一致”的章或手写并签字确认。 | ||
| 9)符合性声明 | ||
| 文件名称 | 文号 |
| 关于第一类医疗器械备案有关事项的公告 | 局令26号 |
| 关于发布第一类医疗器械产品目录的通告 | 局令8号 |
| 国家食品药品监管总局关于印发体外诊断试剂分类子目录的通知 | 食药监械管〔2013〕242号 |
| 国家食品药品监管总局关于实施《医疗器械注册管理办法》和《体外诊断试剂注册管理办法》有关事项的通知 | 食药监械管〔2014〕144号 |
| 《医疗器械生产监督管理办法》 | 局令7号 |
| 《医疗器械说明书和标签管理规定》 | 局令6号 |
| 《医疗器械注册管理办法》 | 局令4号 |
| 关于实施第一类医疗器械备案有关事项的通知 | 总局食药监办械管〔2014〕174号 |
| 一类医疗器械产品变更备案 | 一类医疗器械备案产品技术要求资料编写服务 |
| 一类医疗器械备案资料有关临床评价资料编写 | 一类医疗器械风险分析报告编写服务 |
| 一类医疗器械自检报告编写服务 | 一类医疗器械生产备案代理申请服务 |

未办理成功可全额退款
明码标价无任何隐性消费
签订合同,开具正规发票
一个项目配多名服务人员
客服二十四小时全天在线

业务咨询一
对接项目、沟通细节
获取落地解决方案

业务咨询二
询价、咨询流程请留言
第一时间回复您
周一至周日 9:00-21:00





