网站导航
13673698348


来自:九游服务医疗 发布时间:2024-02-26 浏览 :614次
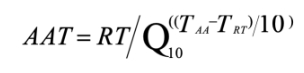
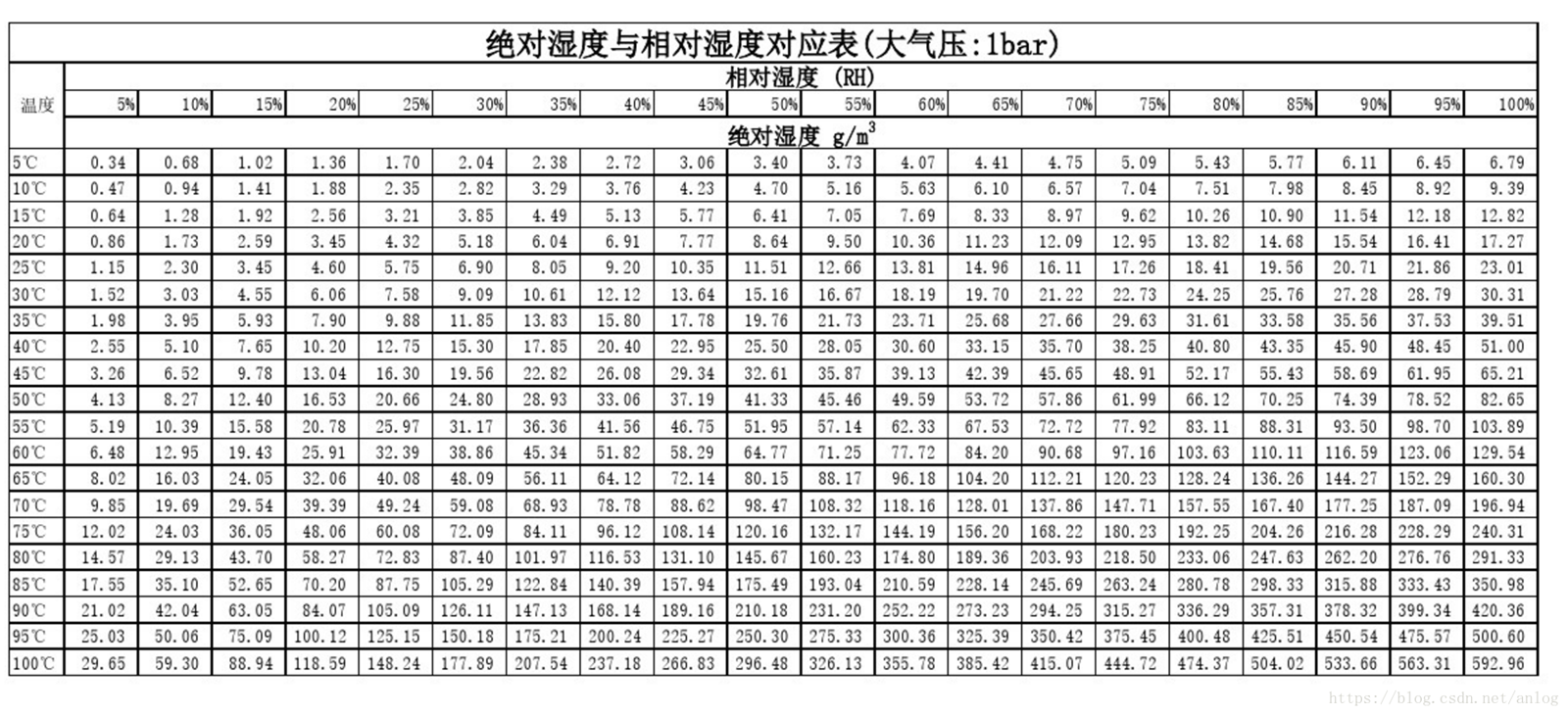

未办理成功可全额退款
明码标价无任何隐性消费
签订合同,开具正规发票
一个项目配多名服务人员
客服二十四小时全天在线

业务咨询一
对接项目、沟通细节
获取落地解决方案

业务咨询二
询价、咨询流程请留言
第一时间回复您
周一至周日 9:00-21:00









